Today I performed the following protocol for DNEasy extraction.
- Vortexed Samples to mix up lysed tissue
- Allowed them to sit for 10 minutes while I made up a gel.
- Added 200 ul Buffer AL with EtOH from the 96 Well Kit
- Added 200 ul 100% EtOH
- This was a mistake as the 96 well kit premixes them and the singles kit does not. We ended up using a 75% EtOH, 25% Buffer AL Solution.
- Vortexed thoroughly
- Pipetted into a column
- Centrifuged at 6000 g for 1 minute
- Discarded collection tube
- Added 500 ul AW1 for wash and new collection tube
- Centrifuge at 6000 g for 1 minute
- Discarded collection tube
- Added 500 ul AW2 for wash and new collection tube
- Centrifuged at 10,000 g for 6 minutes
- Discarded collection tube
- Placed column in labelled 1.5 ml tube
- Added 200 ul AE elution to column
- Incubated 1 minute at room temp
- Centrifuged at 6000 g for 1 minute
- Repeated steps 16, 17, 18.
- Discarded column and stored at room temp.
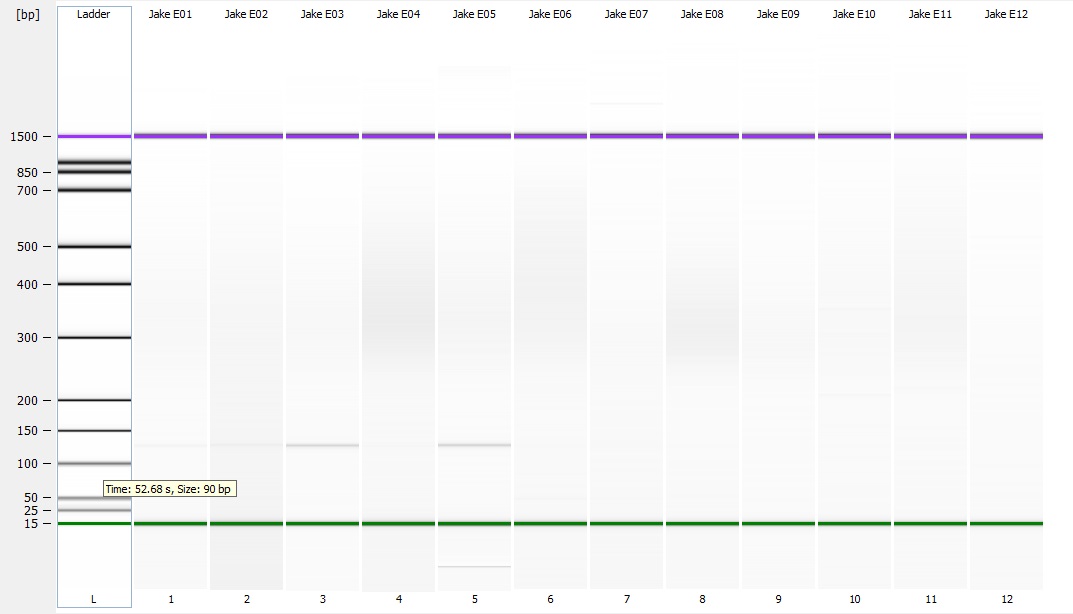
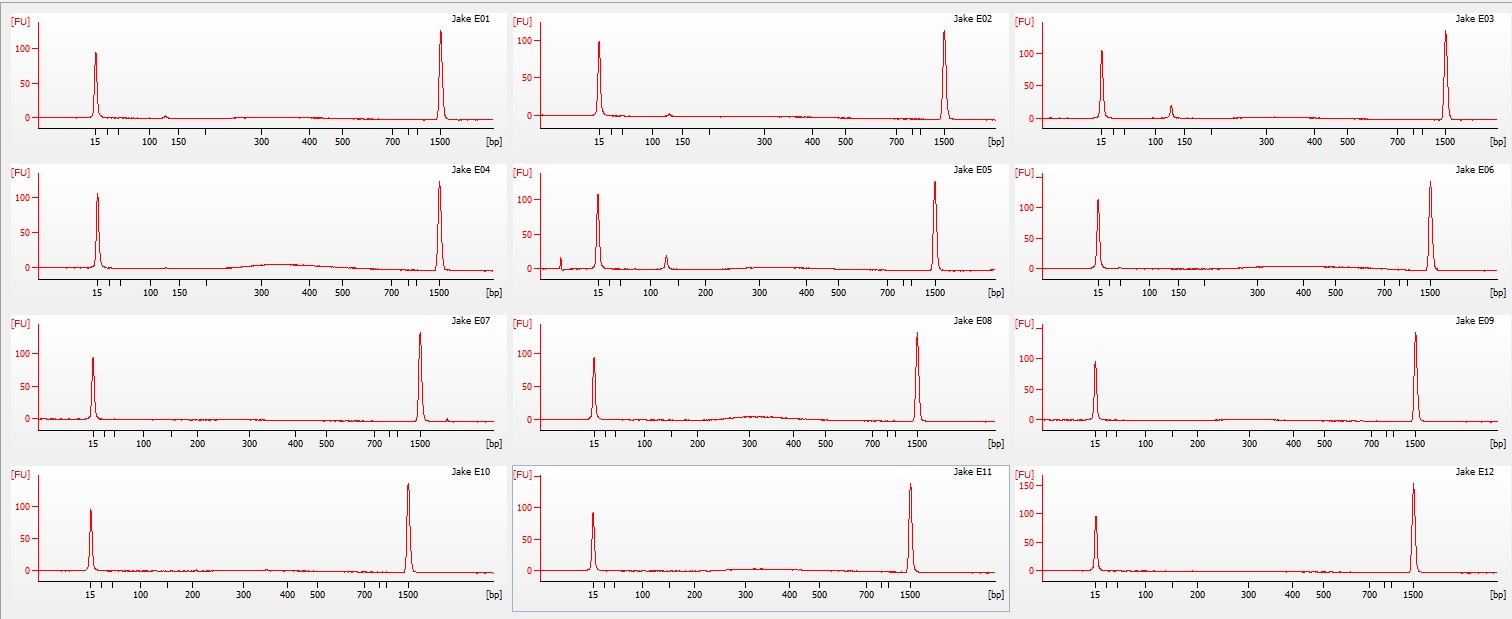
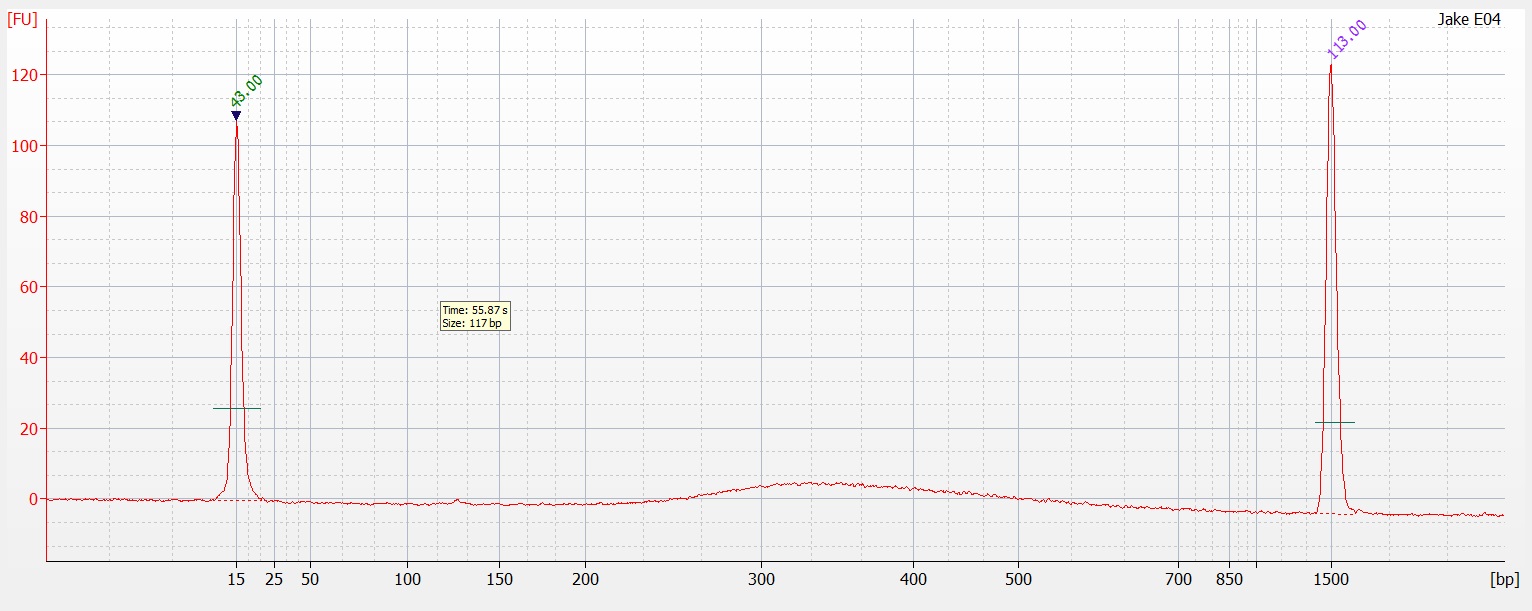
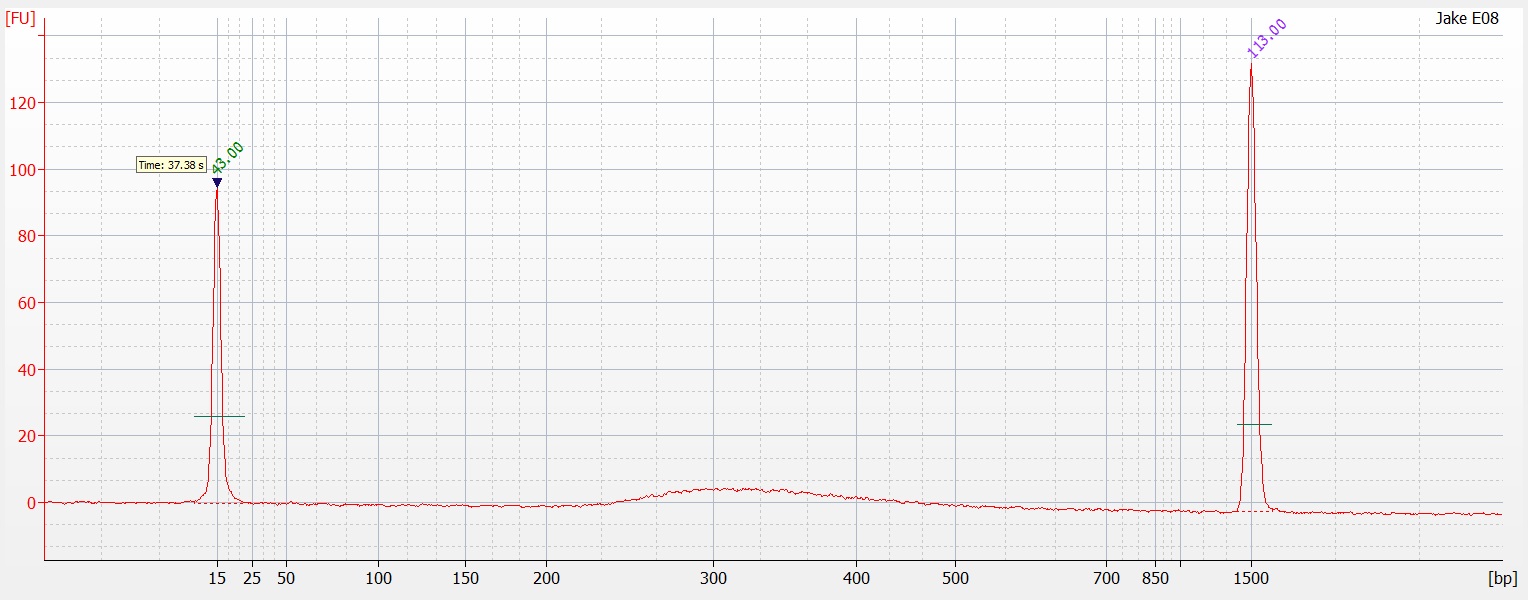
Following the completion of the DNeasy extraction I ran a gel with both sets of isolations.
0.9% Gel:
50 ml 1X Low TAE
0.45 g Agarose
Microwaved gel for 4 minutes. Allowed to cool for 10 after thoroughly dissolved. Poured gel smoothly. Allowed to set for 30-45 minutes.
Loaded wells with
10 ul 100 bp Ladder
25 ul DNA with 2.5 ul Loading dye.
Wells organized like:
| Well | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Technique | DNAzol | DNEasy | ||||||||||
| Sample | Ladder | 1N9-12 18 | 1N9-12 19 | 1N9-12 20 | 1N9-12 21 | 1N9-12 18 | 1N9-12 19 | 1N9-12 20 | 1N9-12 21 | Empty | Ladder | Empty |
 |
| Gel comparing DNAzol to DNEasy |
Moving forward, we should consider doing the 96 well extraction again with smaller tissue sizes, less incubation time, and possible the 75/25 mix of the AL solution.